Pipeline Setup
Overview
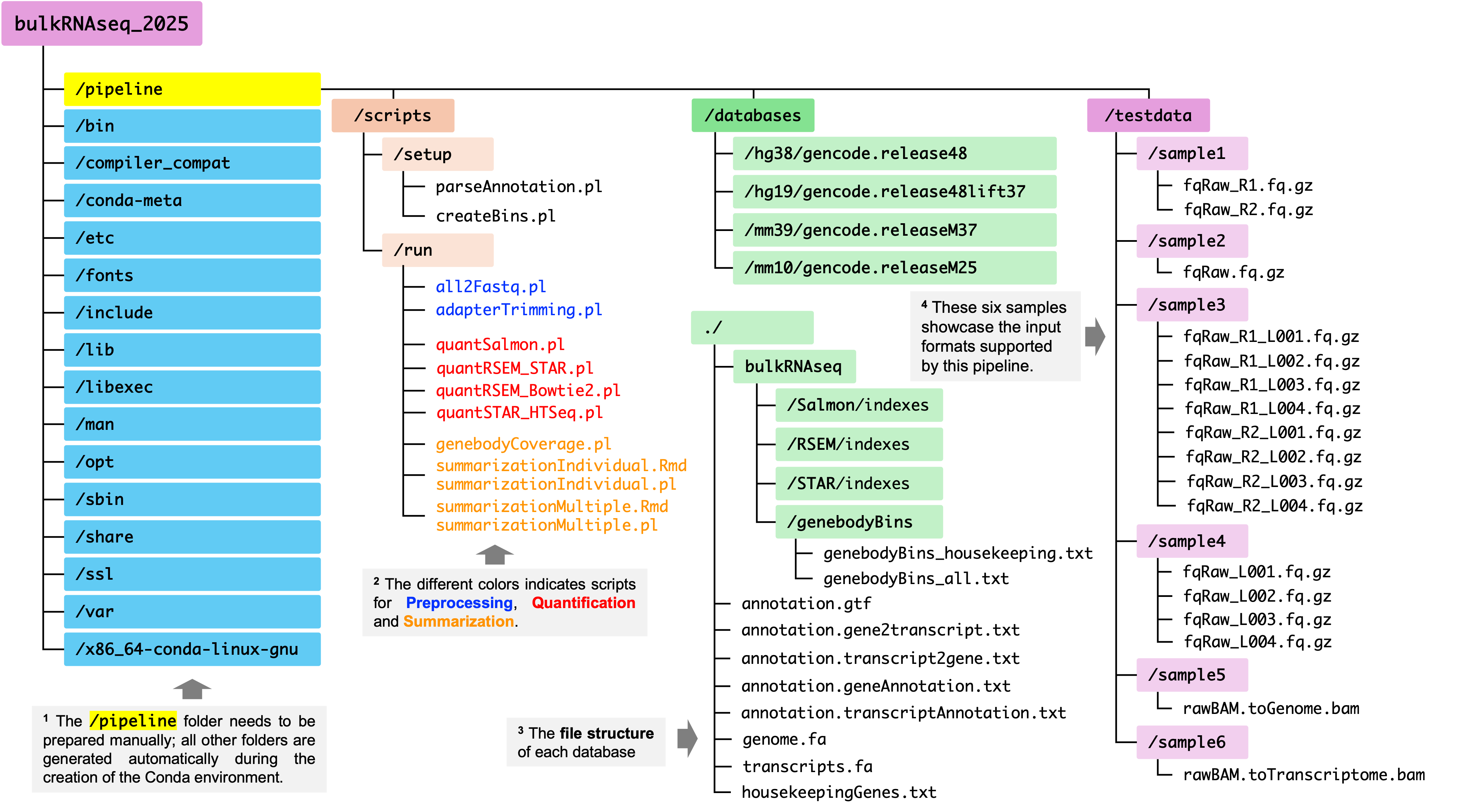
This pipeline can be set up in one single conda environment. The figure below illustrates the file structure of this pipeline which contains three conponents:
-
Software:
Inlcudes all requried tools and dependencies, which are installed during the creation of conda environment. Additionally, several in-house scripts need to be manually deposited to
/path-of-your-conda-env/pipeline/scripts. -
Databases:
For each refrence genome assembly, a dedicated folder is created to store all necessary files for the pipeline, such indexing files for alignment and quantification, and bin list of genes for quality control analysis.
-
Test data
They are curated for three purposes: 1) Showcasing the input formats accepted by this pipeline; 2) Benchmrking pipeline performance (e.g., Run Time, CPU Time, Max Memory Usage); and 3) For users who don’t have their own bulk RNA-seq data, they can use the test data to walk through the pipeline.
The setup of this pipeline consists of three main sections:
-
Software Installation
Now the computational environment for this pipeline is managed within a single conda environment. In this section, you will create a conda enviroment for this pipeline, install all required tools and dependencies, and deposite the scripts of this pipeline to the appropriate directory. For step-by-step instructions, please refer to: Software Installation.
-
Database Preparation
In addition to software, the pipeline requires various reference files for alignment, quantification, and quality control. These files include indexed genome files, gene body bin lists, gene/transcript annotation files, and more. In this pipeline, we organize these files in a per reference genome assembly, per folder manner: each reference genome assembly has its own folder containing all required files for every tool and analysis. For detailed guidance, please see: Database Preparation.
-
Test Data collection
This step is required ONLY WHEN you do not have your own data to walk through this pipeline. We always encourage users to test and explore this pipeline using their own data, if available.
Before you start
We have already set up this pipeline in a dedicated conda environment, which can be activated using the commands below:
module load conda3/202402 # version:24.1.2
conda activate /research_jude/rgs01_jude/groups/yu3grp/projects/software_JY/yu3grp/conda_env/bulkRNAseq_2025
Within this conda environment, we have pre-built databases for the four most commonly used reference genome assemblies of human and mouse: hg38, hg19, mm39 and mm10.
| Genome | GENCODE release | Release date | Ensembl release | Path |
|---|---|---|---|---|
| hg38/GRCh38.p14 | v48 | 05.2025 | v114 | /research_jude/rgs01_jude/groups/yu3grp/projects/software_JY/yu3grp/yulab_databases/references/hg38/gencode.release48 |
| hg19/GRCh37.p13 | v48lift37* | 05.2025 | v114 | /research_jude/rgs01_jude/groups/yu3grp/projects/software_JY/yu3grp/yulab_databases/references/hg19/gencode.release48 |
| mm39/GRCm39 | vM37 | 05.2025 | v114 | /research_jude/rgs01_jude/groups/yu3grp/projects/software_JY/yu3grp/yulab_databases/references/mm39/gencode.releaseM37 |
| mm10/GRCm38.p6 | vM25 | 04.2020** | v100 | /research_jude/rgs01_jude/groups/yu3grp/projects/software_JY/yu3grp/yulab_databases/references/mm10/gencode.releaseM25 |
*: Updates for the hg19/GRCh37 genome assembly ceased in 2013. However, gene annotations continue to be maintained by mapping the comprehensive gene annotations originally created for the GRCh38/hg38 reference chromosomes onto GRCh37 primary assembly using gencode-backmap.
**: Updates for both the mm10/GRCm38 genome assembly and its gene annotations ended in 2019.
You should set up your own pipeline only when:
- You are unable to access to the conda environment above; Or,
- You require a reference genome assembly other than the four pre-built ones.