Part I: Software Installation
As mentioned before, we have manged to install and maintain all required tools and dependencies in one single conda environment. To set up this computational environment:
-
Make sure conda is already available in your case. If not, please install it following this guidance.
module load conda/202402 # version 24.1.2NOTE: Though we always encourage the users to create the conda environment and install the required tools and dependencies MANUALLY, we do provide the
environment.ymlfile (available here) so that the users can directly check the tools and dependencies and easily create the conda environment using:conda env create -f environment.yml -n bulkRNAseq_2025 -p /your-own-path/bulkRNAseq_2025 -
Create a conda environment for this pipeline:
conda create \ --name bulkRNAseq_2025 \ --prefix /research_jude/rgs01_jude/groups/yu3grp/projects/software_JY/yu3grp/conda_env/bulkRNAseq_2025 \ python=3.9 r-base=4.4- Please modify the
--prefixaccordingly. If ignore it, the conda enviroment will be installed in the default directory. - Make sure
python=3.9and **r-base=4.4**are specified. There might be some compatiable issues for the default python and R version.
- Please modify the
-
Install key software and dependencies:
## activate the conda env conda activate /research_jude/rgs01_jude/groups/yu3grp/projects/software_JY/yu3grp/conda_env/bulkRNAseq_2025 # please modify accordingly ## install tools from bioconda channel conda install -c bioconda rseqc=5.0.4 fastp=1.0.1 biobambam=2.0.185 samtools=1.22.1 fastqc=0.12.1 bedtools=2.31.1 bowtie2=2.5.4 rsem=1.3.3 star=2.7.11b salmon=1.10.3 cutadapt=5.1 htseq=2.0.9 ucsc-genepredtobed=482 ucsc-gtftogenepred=482 ## install dependencies from conda-forge conda install -c conda-forge pandoc=3.7.0.2 ## install R packages from r channel conda install -c r r-rmarkdown=2.29 r-ggplot2=3.5.2 r-dplyr=1.1.4 r-envstats=3.1.0 r-kableextra=1.4.0 r-rjson=0.2.23 r-cowplot=1.2.0 r-plotly=4.11.0 ## deactivate conda env conda deactivateTroubleshooting: Some of the tools of the specified versions can not be installed in your case?
One of the major challenges in setting up this pipeline was identifying compatible tools and their versions. The versions specified above have been thoroughly tested in Red Hat Linux and Ubuntu and work well together. However, compatibility is not guaranteed in all environments, especially if you are using a different OS or setting up the pipeline at a date way later than Sept. 2025. If you encounter issues installing a specific version, you can try omitting the version tag and using the tool name only. Conda, in most cases, can automatically select a competible version for you. If you continue to experience difficulites, please feel free to reach out to us via Qingfei.Pan@stjude.org for further assistance.
-
Deposite the home-made scripts
In additon to the tools and dependencies installed above, this pipeline relies on several in-house scripts to set up and run this pipeline. All these scripts are available here. You may use any preferred method to download them, but we recommend the following approach:
- Copy this link: https://github.com/jyyulab/bulkRNAseq_quantification_pipeline/tree/main/scripts
- Visit this directroty downloading tool at: https://download-directory.github.io/
- Paste the copied link into the search box, and press Enter.
- Unzip the downloaded file and save them to your conda environment at:
/path-to-your-conda/pipeline/scripts.
NOTE: Since this pipeline documantation is based on a specific structure (as shown in the figure below), to avoid any unnessary confusion, please DO NOT change the path to deposite these script files unless you have a specific reason to do so. For reference, in our pre-built conda environment, these script files should be deposited to:
/research_jude/rgs01_jude/groups/yu3grp/projects/software_JY/yu3grp/conda_env/bulkRNAseq_2025/pipeline/scripts, as shown in the figure blow.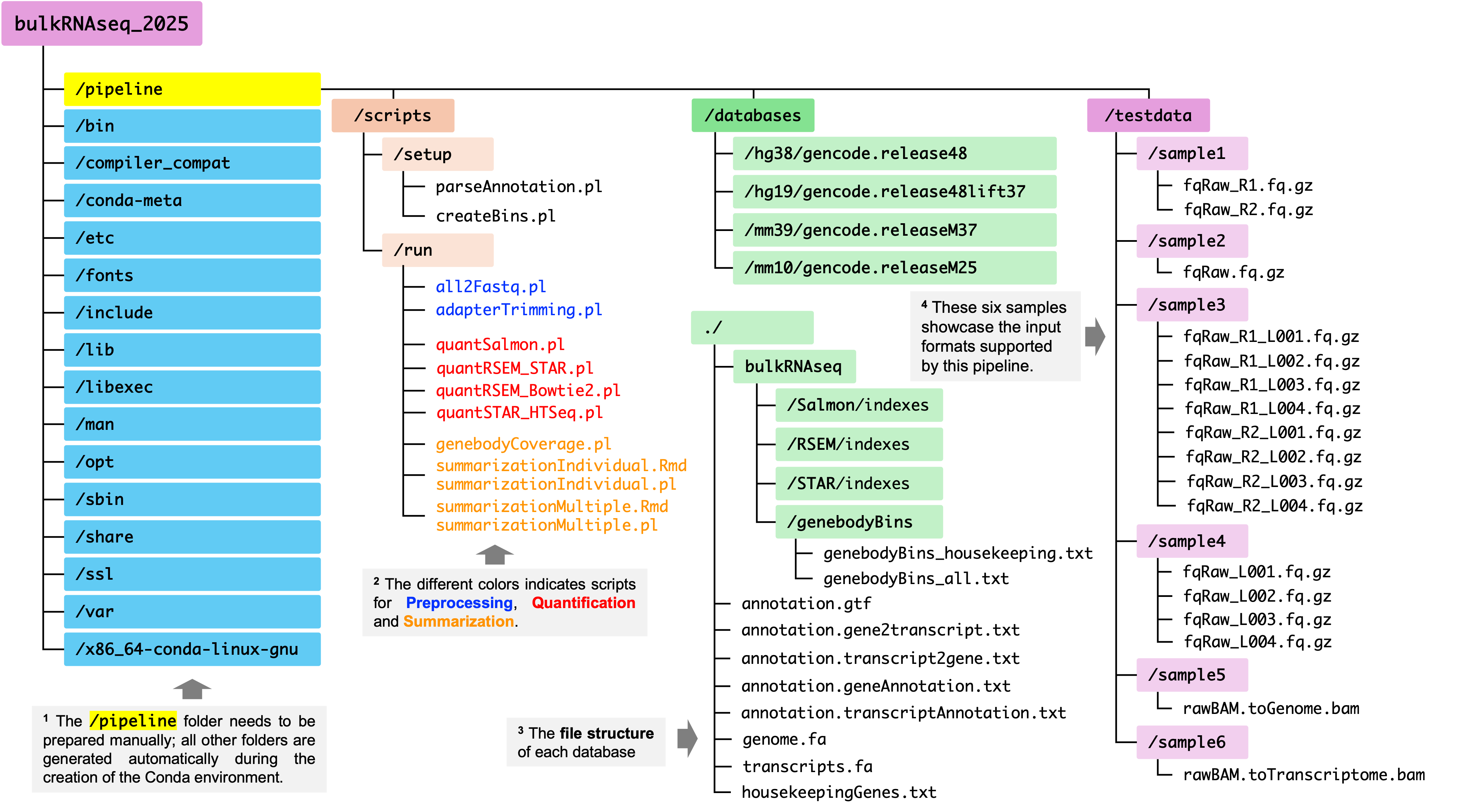
After completing the four steps above, you should see the
/pipline/scriptsfolder in your conda environment. Inside this folder, there are two subdirectories:setup: Contains two scripts used to setting up the databases for reference genome assemnbliesrun: Contains 11 scripts that execute the three main stages of the pipeline: Preprocessing, Quantification and Summarization.